Mass spectrometry is a technology which is widely used in most scientific disciplines that require accurate and precise measurement of elemental and molecular components. Its use in the pharmaceutical sector is often associated with the Drug Discovery and Development process. The primordial step to analyze a sample is to perform a sample treatment to enable its study. It really requires know-how to obtain good quality results. The following tips and tricks can help you to rapidly select a protein purification strategy and to anticipate some possible problems.
Mass spectrometry is an instrumental method for a device for identifying the kinds of particles present in a given substance: the particles are ionized and beamed through an electromagnetic field and the manner in which they are defected is indicative of their mass and, thus, their identity and characterized their chemical structure. The principle is the separation of gas-phase ions (charged molecules) of the compound in function of their mass-to-charge ratio (m/z).
This technology allows you to :
- determine the precise molecular weight of proteins (for confirmation of sample identity, determination of sample purity and amino acid composition, identification of post-translational modifications and di-sulfide bonds)
- sequence (for protein sequence confirmation, for peptide characterization, for protein identification by comparing wit databases)
- follow reactions (enzymatic reactions, chemical modifications and protein digestion)
Biopharmaceuticals are more complex than small molecules due to their biotechnological synthesis process which induces biological and chemical impurities and protein heterogeneity. This observed heterogeneity can be the consequence of chemically modifies forms, interaction with the matrix, post-translational modifications or different folding states.
The sample to study can have different origins. It can be simple sample collection from animals or humans or recombinant protein production in prokaryotic or eukaryotic cells (see Fig. 1).

If you want to study an isolated protein, several purification strategies can be employed based on the protein characteristics.
At the production step, the recombinant protein can be produced either with a tag or without a tag. The tag can simplify the purification process and sometimes, depending on the nature of the tag, can enhance protein solubility.
#1: Protein purification step
The protein purification process is typically divided in 4 steps:
- Lysate clarification, the sample preparation and the protein extraction
- Protein capture which corresponds to the protein isolation, extraction and stabilization
- Intermediate purification step which permits to remove bulk impurities
- Polishing purification step which corresponds to the achievement of final high level purity
The number of steps can be increased in order to obtain a purer protein.
For each step, you need to choose an adapted strategy to purify your protein of interest. You can find below a table summarizing the best strategy to use by taking into account the protein characteristics (see Table 1). In front of each technique is associated are my recommendations concerning the purification step in which you can use it.

#2: But sample treatment is not only a purification step…
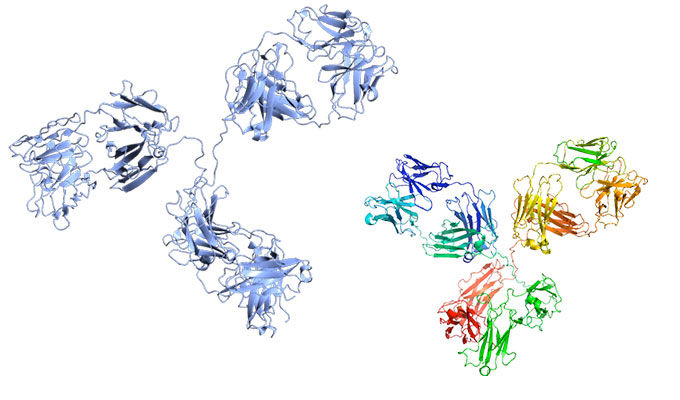
2 major strategies exist. The first one is named “top-down proteomics” and is the most useful for single proteins or relatively simple mixture (See Fig. 2). It’s a powerful strategy to distinguish sequence variants or to decipher combinatorial modifications on a protein. Nevertheless, it’s not suitable for most biomedical applications due to the incapacity to easily analyze complex biological samples, like in proteome studies, and also due to the requirement of highly efficient and specialized instrumentation. Moreover, the interpretation of the Data is quite complex and lacks automatization.

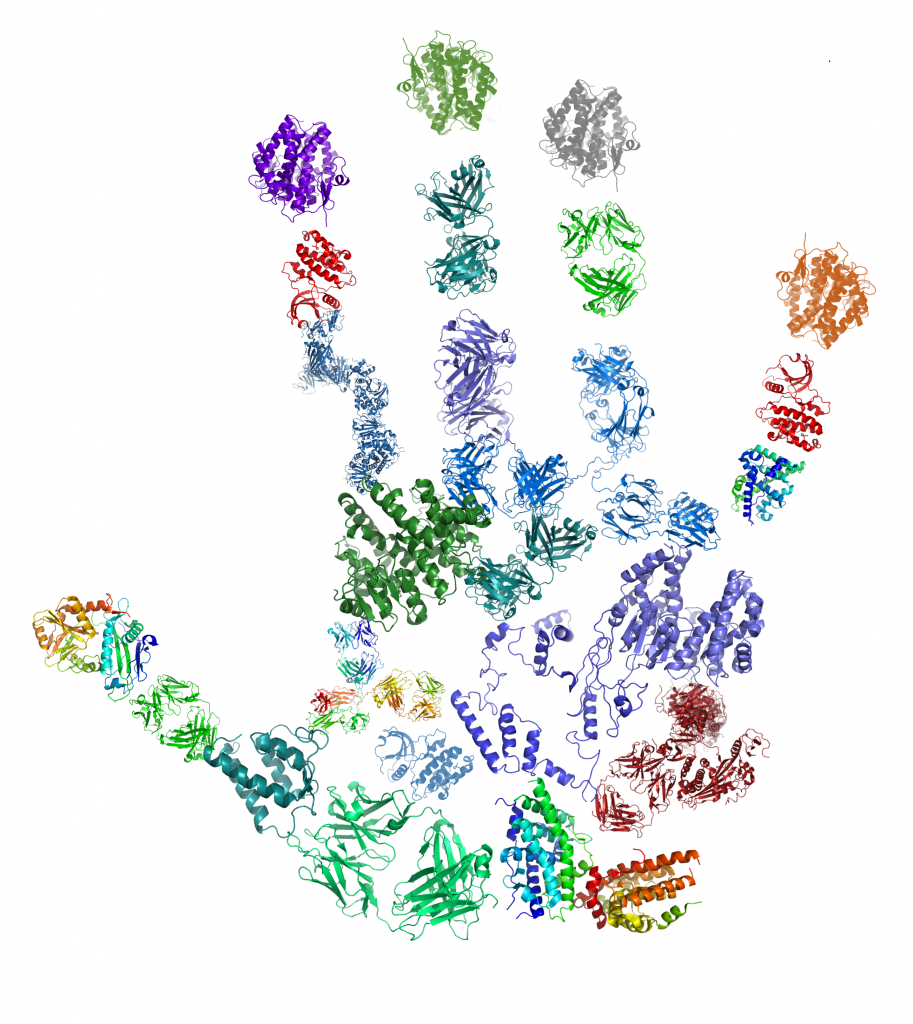
The second one is named the “bottom-up proteomics” which can be applied on isolated protein(s) and also on crude extract and which requires a protein digestion step (see Fig. 3).

One of the most important step is to choose the right MS-grade protease for your application (see Table 2 below with cleavage specificity of classical proteases). Please note, that the peptide / protein identification is improved when same samples are treated separately with different proteases (like combination of Trypsin / Lys-C / Chymotrypsin digestions).
This strategy has a lot of advantages like the easily automated data acquisition and the fragmentation of tryptic peptides which are well understood. Moreover, a lot of efficient software is available on the market for the analysis and for some of them free access exists. Concerning whole proteome analysis, separation of peptides allows to create less complex subsets for MS analysis which is far easier than for proteins. Naturally, there are also any disadvantages like the loss of relationship between peptides and the original protein and the complexification of the mixture by generating 20-100 x more fragments.
These two strategies enable you to quantify, identify and characterize post-translational modifications from one protein to the entire proteome.
#3: Tricks to know for obtaining quality data
Sample treatment is not an easy thing and really requires know-how to obtain satisfying and quality results which can easily be interpreted.
For example, some compounds must be avoided during sample preparation:
- Salts which cause adduct formation
- Detergents and polymers which form polymer series and compete with the sample and which can also suppress ionization
- Non-volatile organic solvent (DMSO, DMF…) which interfere with ion desolvation
- Strong acids (like TFA) which can suppress signals from the analyte of interest
- Keratins which can monopolize the spectrometer
In most of cases, an additional purification step can eliminate these compounds with extensive washing steps or desalting purification but… the simplest strategy is to not use them!
Interested in additional tips and tricks for protein production and protein purification?
- How to choose the perfect buffer to get a pure, stabilised functional protein
- Gel Filtration – Which column to choose for your size exclusion purification?
- How to easily purify your protein with adapted magnetic beads…
- Why will mixed mode resins revolutionise R&D scale mAb production?
- How to easily obtain a highly purified untagged recombinant protein
Don’t hesitate to contact me for additional recommendations, or to define the most adapted protocol for the treatment of your sample before MS analysis. Just leave your comments below!